

A Professora de Farmacologia da FMUL, Cristina Sampaio e um grupo internacional de cientistas desenvolveram um novo sistema de estadiamento da Doença de Huntington que cobre a totalidade do curso da doença, desde o nascimento até à morte. O novo sistema identifica os casos de doença de Huntington com base no teste genético e permite standardizar as fases iniciais da doença, o que vai facilitar a condução de ensaios clínicos neste período.
A Doença de Huntington (DH) é uma doença neurodegenerativa genética. Clinicamente cursa com alterações do movimento, particularmente movimentos involuntários do tipo coreia, deterioração cognitiva e alterações do comportamento. Antes de serem detetados sintomas, a doença evolui durante décadas, uma das alterações mais características é atrofia do caudado e do putamen facilmente detetável na ressonância nuclear magnética do crânio. Esta atrofia começa 10 a 15 anos antes dos sintomas.
Depois do aparecimento dos sintomas a doença evolui para a morte em cerca de 15 anos. O novo sistema de estadiamento tem alguma analogia com o estadiamento da doença oncológica. No novo sistema são considerados 4 estádios de progressão (do 0 ao 3, sendo o 3 o mais elevado).
O estudo publicado pela The Lancet Neurology foi conduzido no âmbito do consórcio “Huntington Disease Regulatory Science Consortium – HD-RSC” que faz parte do C-Path Institute.
O sistema de estadiamento apresentado neste estudo vai facilitar o desenvolvimento de ensaios clínicos em estádios precoces, quando os sintomas não são ainda detetáveis, o que se julga aumentar a probabilidade de sucesso de medicamentos que visam retardar a progressão da doença. De momento, ainda não há nenhum tratamento aprovado que modifique o curso da doença.
O novo sistema de estadiamento foi desenvolvido para uso no contexto de investigação clínica da Doença de Huntington, a sua translação para a prática clínica poderá acontecer mais tarde.

Porque surgiu o interesse em estudar mais esta doença?
Cristina Sampaio: Bom, esta é uma pergunta que obrigaria a uma resenha autobiográfica. Como não há espaço para tanto, direi que enquanto estudante de Medicina tive o privilégio de ser convidada pelo Professor Castro Caldas para trabalhar com ele em vários projetos científicos, a maioria dos quais relacionados com a Doença de Parkinson. Ora a doença de Parkinson é um dos mais representativos elementos de um grupo chamado - Doenças do movimento. A Doença de Huntington (DH) é outro elemento desse mesmo grupo de doenças. Ao especializar-me em doenças do movimento, e em metodologias da investigação clínica, juntei um conjunto de aptidões que serão a razão para ter sido convidada para dirigir o departamento clínico da CHDI Foundation. Esta é uma fundação americana, sem fins lucrativos, cuja missão é facilitar o desenvolvimento de terapias suscetíveis de alterar positivamente o curso da doença. Portanto, o interesse inicial foi circunstancial, mas terei que acrescentar que a doença de Huntington tem uma base biológica fascinante; os mecanismos associados à mutação causal são complexos e a doença tem um curso muito prolongado (várias décadas), a fase sintomática só acontece ao fim de algumas décadas de vida. Tentar compreender o que acontece desde o nascimento até a manifestação dos sintomas é um desafio intelectual que me atrai.
No mundo e em Portugal, quantas pessoas são afetadas com a doença de Huntington?
Cristina Sampaio: Costumamos dizer que a DH é a menos rara das doenças raras. Estima-se que a prevalência seja cerca de 14 em 100000 nas populações de origem europeia. Portugal deverá ter pelo menos cerca de 1400 pessoas com DH. Quando se fala de prevalência da DH importa esclarecer que na quase totalidade dos estudos epidemiológicos só contam como casos, as pessoas que têm sintomas. Ora como disse acima, os sintomas aparecem por volta dos 40 anos de idade; assim, há muitos indivíduos que são portadores da mutação, mas que na altura em que os estudos são feitos ainda não têm sintomas, mas já estão doentes. Estes indivíduos portadores sem sintomas, não têm sido contados. Como a doença é herdada de forma autossómica dominante, os filhos de um indivíduo portador da mutação tem 50% de probabilidade de também serem portadores. Tendo em conta o tipo de hereditariedade e alguns outros fatores, estima-se que por cada caso sintomático haja 3 a 5 casos ainda sem sintomas. Usando este tipo de cálculo em Portugal deverá haver entre 4200 a 7000 indivíduos portadores da mutação que causa a DH.
Há países com maior predominância desta doença?
Cristina Sampaio: A distribuição geográfica da DH é bastante assimétrica porque a mutação original no gene causal ocorreu numa pessoa de etnia caucasiana e tem-se propagado nas populações dessa mesma etnia. Sabe-se que existem mutações espontâneas, ou seja, que não foram herdadas, mas são muito raras. Há DH em outros contextos étnicos que não o caucasiano, mas as estimativas que se conhecem sugerem que a prevalência noutras etnias pode ser cerca de 10 vezes inferior à dos países europeus, e também é variável de etnia para etnia.
E interessante notar que existem pequenos enclaves geográficos, maioritariamente, localizadas na América do Sul (o mais conhecido situa-se na Venezuela à volta do lago Maracaibo) em que a prevalência pode chegar a ser 1000 vezes a dos países europeus. Estes clusters resultaram de um indivíduo portador da mutação ter chegado a estas regiões, alguns séculos atrás, vindo da Europa (fala-se sempre de um possível marinheiro português ou espanhol!) e ter constituído família, perpetuando assim a transmissão da mutação num contexto social em que os casamentos consanguíneos são frequentes e, por razões de geografia e pobreza, a mobilidade é limitada. Estes últimos condicionamentos multiplicam a probabilidade de transmissão da mutação fazendo crescer os casos de doença a uma taxa muito elevada o que justifica a prevalência inusitada.

Irá ser possível combater a doença com os novos fármacos que estão em desenvolvimento?
Cristina Sampaio: A maioria das pessoas que trabalham neste campo estão convencidas que se verão os progressos, como por exemplo, um ou mais medicamentos poderão ser licenciados num prazo razoável. Na minha opinião esse prazo é ainda de 5 a 10 anos. Tenho colegas que são mais otimistas, acreditam que antes de 5 anos haverá um novo medicamento licenciado.
Há muitos projetos em curso. A compreensão dos mecanismos biológicos da DH associado ao desenvolvimento de biotecnologia que permitiu o avanço de medicamentos baseados no RNA ou mesmo a edição do genoma (CRISPR, Talenos, "Zinc finger proteins") permite a criação de intervenções dirigidas a alvos muito específicos, o que pode prenunciar alguns sucessos semelhantes aos que se obtiveram em oncologia.
Quais as principais vantagens deste novo sistema de estadiamento da Doença de Huntington?
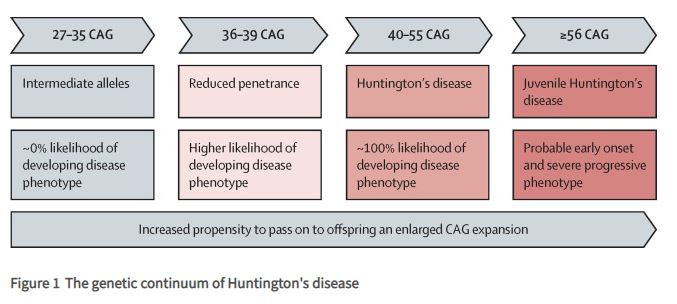
Cristina Sampaio: O sistema de estadiamento (HD-ISS - Huntington Disease Integrated Staging System) que publicamos recentemente é um sistema que permite descrever de forma integrada a progressão da DH desde o nascimento até à morte; uma das características mais importantes reside na definição dos casos de doença pela presença da mutação causal (expansão do número de tripletos CAG acima de 39 no gene Htt, que esta localizado no cromossoma 4); tradicionalmente, os casos são definidos pela presença de sintomas específicos, o que só acontece tardiamente.
O estadiamento com o HD-ISS permite classificar de uma forma precisa as pessoas com HD em 4 estádios (os limiares para cada teste usado como marcadores de estadio foram calculados com base em dados de milhares de indivíduos, e são referenciados a população controlo). O estádio 0 representa indivíduos que são portadores da mutação, mas não têm nenhuma alteração biológica detetável com os métodos existentes; estádio 1 – indivíduos que além de serem portadores da mutação, têm atrofia dos núcleos do estriado (áreas subcorticais do cérebro) o que assinala que o processo neurodegenerativo já a se iniciou; estádio 2 – indivíduos que têm as condições de 0 e1 e além disso já tem alguns sintomas motores ou cognitivos avaliados pelas escalas conhecidas por TMS ou SDMT; e estádio 3 – indivíduos que tem as características para 0,1 e 2 e além disso tem incapacidade funcional avaliada pelas escalas conhecidas como TFC e IS.
A classificação precisa num determinado estádio facilita muito a definição de populações para inclusão em ensaios clínicos, o que é particularmente importante para se desenvolver desenhos e métodos de avaliação que permitam fazer ensaios em populações precoces, como por exemplo, populações que não têm qualquer sintoma (estádios 0 ou 1). Até agora, estas populações nunca foram estudadas em ensaios clínicos por falta de metodologias apropriadas e de um caminho regulamentar que seja reconhecido por agências como a EMA ou FDA. Estamos convencidos que o sistema de estadiamento é um primeiro importante instrumento para permitir estes ensaios. O HD-ISS é também um instrumento de comunicação entre os diferentes parceiros envolvidos no processo de desenvolvimento de medicamentos: profissionais de saúde, pessoas com DH, investidores, reguladores, companhias de seguros, etc. Digamos que permite criar uma linguagem comum o que facilita muito a obtenção de compromissos.
Quais são a especialidades médicas que intervêm no tratamento da doença de Huntington?
Cristina Sampaio: O acompanhamento e tratamento de cada caso de DH pode (e deve) envolver uma multitude de especialidades e de apoios profissionais. Na fase sintomática da doença a especialidade médica que tende a gerir o caso é a Neurologia, não só porque a DH é uma doença neurológica, mas também porque os sintomas mais frequentes são motores ou cognitivos. Acontece que muitas pessoas com DH também têm alterações do comportamento e do humor, o que por vezes determina que o primeiro especialista que procuram seja um psiquiatra. À medida que a doença progride e se torna mais incapacitante os apoios da terapia ocupacional, da fisioterapia, da assistência social são fundamentais. Atualmente, só há dois medicamentos aprovados para o tratamento de um dos sintomas motores da DH – a coreia (movimentos involuntários dos vários segmentos do corpo). Esses medicamentos são a tetrabenazina e a deutetrabenazina. No manejo da DH também se usam outros medicamentos, por exemplo os aprovados para o tratamento de depressão e da ansiedade.
De notar que antes de aparecerem os sintomas, a maioria dos indivíduos sabe que está em risco de ser portador e pode considerar fazer um teste genético para saber se o é. Neste contexto os especialistas de maior relevância são os geneticistas. A decisão de fazer este tipo de teste é bastante complicada do ponto de vista pessoal e tem importantes consequências para a vida. As consequências não são só do ponto de vista existencial, condicionam também decisões sobre carreira, casamento, filhos e reprodução medicamente assistida. Correntemente a maioria dos indivíduos nestas circunstâncias ainda opta por não fazer o teste (ou simplesmente não tem suficiente informação para tomar tal decisão). Por isso, o uso de diagnóstico genético pré-natal associado a fertilização in vitro é subutilizado.
O desenvolvimento bem-sucedido de um medicamento para DH poderá dar pistas para o tratamento de outras doenças neurológicas?
Cristina Sampaio: “Sim e não", por um lado alguns alvos terapêuticos na DH são muito específicos porque são determinados quer pelo gene causal, quer por outros genes que se descobriu influenciam a ação do gene causal. O gene causal - Htt - não tem um papel importante conhecido noutras doenças neurológicas. Os genes modificadores atuam em mecanismos como o da reparação do DNA que tem um papel em várias doenças conhecidas como doenças com expansão de tripletos – há 7 destas doenças (por exemplo, a atrofia espinocereberal tipo 1, distrofia miotónica). É lógico assumir que se se fizer um avanço no desenvolvimento de um medicamento que afete a instabilidade somática dos genes mutados numa destas doenças, esse medicamento poderá ter impacto não só na DH, mas também nas outras doenças dessa família (doenças com expansão de tripletos).
Por outro lado, há também projetos focados em alvos terapêuticos que podem ter relevância em várias doenças neurológicas degenerativas - estes são alvos que estão no meio da cascada de eventos biológicos que leva a neurodegeneração e, por isso, são menos específicos que os que mencionei acima. Se houver sucesso numa destas linhas de investigação poderá ter repercussão transversalmente em várias doenças.
Finalmente, a DH é vista como um instrumento de aprendizagem, um modelo, para a estratégia de desenvolvimento de medicamentos para doença de Alzheimer ou doença de Parkinson, entre outras. Em todas estas doenças há um período relativamente longo em que o processo neurodegenerativo está a evoluir, mas não há maneira de o detetar porque as pessoas afetadas têm poucos ou nenhuns sintomas. Como já expliquei, este problema na DH está resolvido pela natureza da doença. Na DH é possível fazer um teste genético e saber com um grau de certeza superior a 95% (95% das pessoas que nascem com CAG de 40 terão sintomas durante o seu período de vida, se o número de CAG for 41 ou maior a probabilidade de desenvolverem sintomas é de 99 a 100%) se aquela pessoa tem a doença, o que permite planear os tais ensaios clínicos precoces. Se um destes ensaios for bem-sucedido haverá muita informação útil não só para DH, mas para as outras doenças que mencionei.
Leonel Gomes
Equipa Editorial
